蛋白質の立体構造の決定の流れ
ここでは、NMR を使用して、蛋白質の立体構造をどのようにして決定するかを簡単に示します。
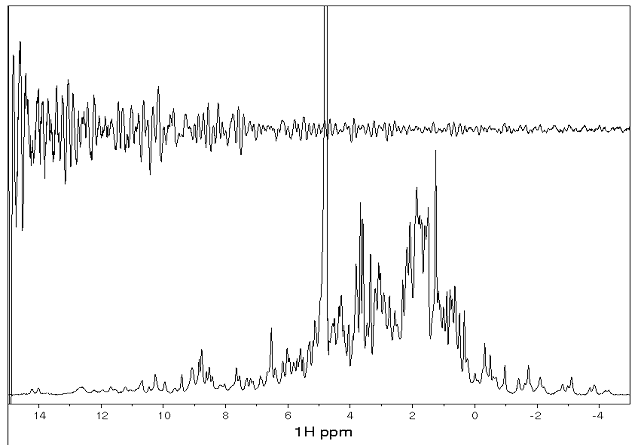
蛋白質の溶液を NMR 装置にセットし、電磁波を与えると下のような FID と呼ばれる信号を検出することができます。これをフーリエ変換したものが、そのすぐ下に示した NMR スペクトルです。この例は一次元のスペクトルです。蛋白質に存在する各水素原子がそれぞれ1本のピークを呈しますので、1000 本以上のピークが重なっており、これでは解析が大変困難です。
|
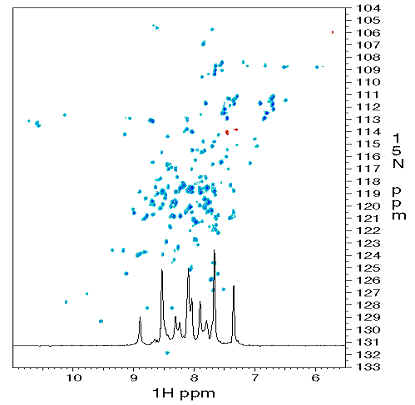
スペクトルを二次元に展開すると、ピークどうしの重なりはかなり緩和されます。下は、各アミノ酸のアミド基における、H と N 核の相関を示すスペクトルで、青いピークのそれぞれが、蛋白質上のどれかのアミノ酸のアミド基に対応します(アミド基の水素と窒素原子核は、それぞれ、緑とピンクで示してある)。したがって、ピークの数は、アミノ酸の数とほぼ同じです(厳密にいうと、アスパラギンやグルタミンの側鎖にあるアミノ基由来のピークも見えています)。
 |
||
 |
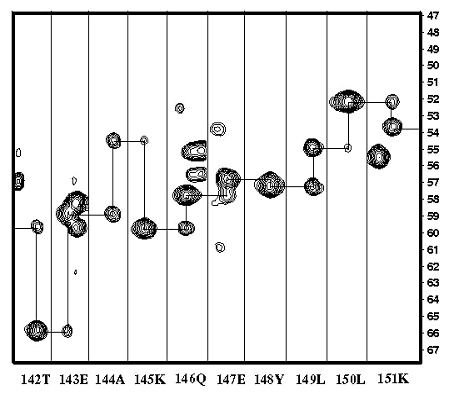
スペクトルの次元数を増やすほど分かりやすくなります。下のスペクトルは、3次元の HNCA スペクトルの一部を切り取って、アミノ酸の配列の順番に並べなおしたものです。連鎖帰属と呼ばれる方法により、それぞれのピークが蛋白質上のどの原子核のペアから由来したのかを決定することが出来ます。この帰属により、各原子核のスピンが磁場の中でどの程度のスピードで歳差運動しているかを求めることができ、化学シフト値(単位は、ppm)の形で登録されます。この化学シフトは、今後の全てのNMRの解析で必要となる、もっとも重要な基本的データとなります。
 |
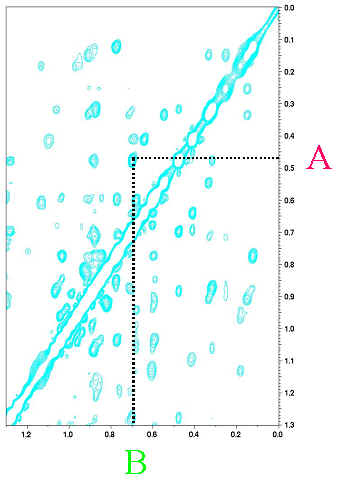
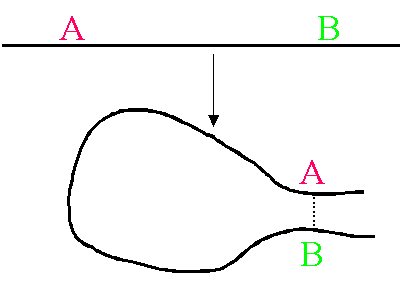
蛋白質が正常に折りたたまれていて、もし、核 A と B が空間的に近い場合、NOESY
と呼ばれるスペクトルを測定すると、両者の間にピークが生じます。先ほどの帰属の結果を参照すると、横軸と縦軸の化学シフト値から、そのピークが核 A と B
に対応することが分かります。これが、立体構造を決定するために重要な距離情報です。核 A と B
は、一次構造上では、離れているかもしれません。しかし、立体構造上では、近くどうしにあるという情報が重要で、これは、距離制限と呼ばれています。
 |
||
 |
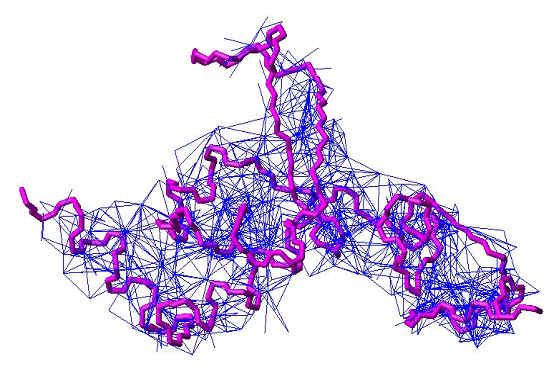
NOE ピークから得られた 1500 個以上の距離情報は、コンピュータに入力され、それら全てを満たす立体構造が試行錯誤的に決定されます。下図の短い青い線は、一つ一つの距離制限を示します。残念ながら、NOE は、核間距離が短い ( 5Å ) 場合にのみ観測されるため、多数の距離制限を集めないと正確で精密な構造が得られません。結合の間の角度(二面角)もスカラー結合定数 J という値から求めることが出来、それらを角度情報として、構造の組み立てに使うことが出来ます。しかし、これも距離情報と同じく近距離に相当する制限情報であることに変わりはありません。
 |
最近、注目され出した遠距離に相当する方向情報の観測法については、追って記述する予定です(次回をお楽しみに)。
